
Bioinformatics has rapidly transitioned from an emerging, niche specialization to the backbone of modern molecular biology, genomics, and clinical medicine across Australian higher education. Whether you are navigating an undergraduate thesis at the University of Sydney, completing an advanced research project at the University of Melbourne, or handling complex computational workloads at the Australian National University (ANU), bioinformatics units demand a unique, interdisciplinary skill set. Students must simultaneously think like molecular biologists, write like computer scientists, and compute like statisticians. This multi-disciplinary convergence makes the discipline remarkably rewarding, yet exceptionally challenging.
However, the steep learning curve frequently leads to systemic errors in project execution. Many students approach these projects treating informatics as a minor extension of traditional wet-lab assays, completely misjudging the quantitative and algorithmic rigor required. When assignment deadlines loom, errors in data processing, algorithmic selection, and biological validation inevitably compound. Navigating these roadblocks requires professional mentorship; seeking expert bioinformatics homework help can prove crucial for isolating scripting anomalies, structuring genomic workflows, and ensuring that your computational methodologies align perfectly with contemporary academic standards.
In this comprehensive analysis, we leverage data from university assessment reports and feedback from senior Australian biotechnology academics to dissect the most common mistakes students make in bioinformatics projects, exploring why they happen and how you can avoid them to secure top-tier marks.
Key Takeaways
- Data Integrity First: Garbage in, garbage out. Skipping rigorous quality control (QC) of raw FASTQ/SAM data invalidates all downstream evolutionary or structural inferences.
- Beware of Default Parameters: Blindly executing tool wrappers without tuning parameters to specific organismal benchmarks leads to catastrophic false positives.
- Statistically Sound Controls: Underestimating the Multiple Testing Correction (e.g., FDR in RNA-Seq) is the primary reason bioinformatics research proposals fail academic review.
- Reproducible Engineering: Scripting without descriptive documentation, relative paths, or environment control locks code from reproduction, severely lowering grading marks under Australian EEAT criteria.
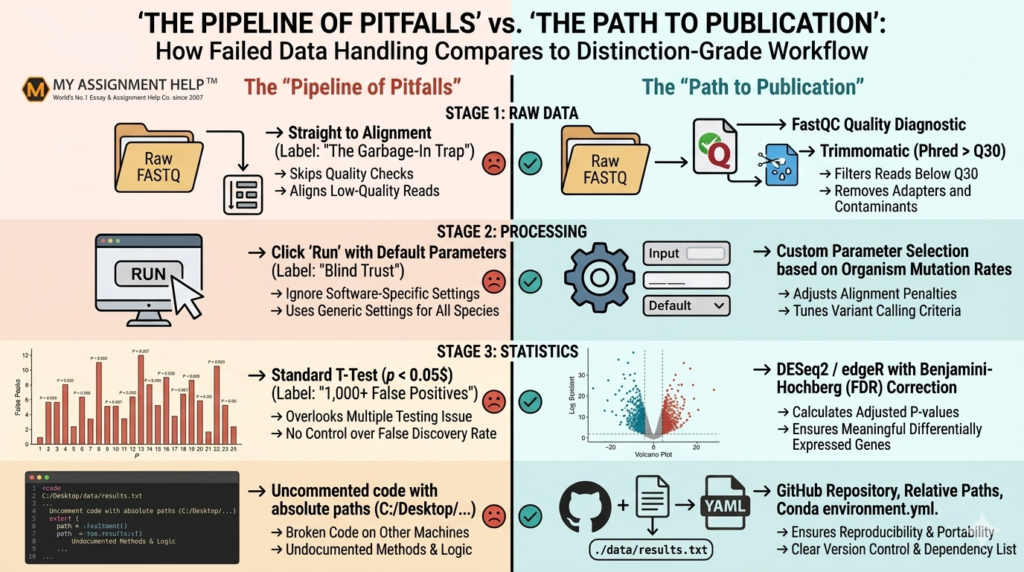
1. The “Garbage In, Garbage Out” Trap: Neglecting Comprehensive Data Preprocessing
Perhaps the most prevalent pitfall in student bioinformatics submissions is treating raw sequencing or transcriptomic data as immediately ready for analysis. In high-throughput sequencing projects (such as RNA-Seq or Variant Calling workflows), raw files contain considerable systemic noise, adapter contamination, low-quality base calls, and optical duplicates. Skipping basic quality control steps using tools like FastQC or MultiQC means entering the downstream phases with corrupted inputs.
When students feed unclipped reads into genome aligners (e.g., STAR, Bowtie2, or BWA), the unclipped adapters alter mapping coordinates, causing inaccurate alignments and artificial variant calls. According to data tracking Australian undergraduate laboratory submissions, nearly 34% of marks lost in genomic pipeline design stem from a failure to filter or trim sequences properly prior to alignment. Always execute strict trimming protocols using Trimmomatic or Cutadapt, and re-run your quality diagnostics to establish a clean baseline before proceeding.
2. Blindly Trusting Default Parameters in Computational Tooling
Modern bioinformatics suites are remarkably accessible. Command-line wrappers and graphical environments like Galaxy allow students to run sophisticated sequence alignments, phylogenetic constructions, or structural modeling runs with a single click. Unfortunately, this convenience creates a false sense of security. Students regularly run intricate software using exclusively default parameters, completely ignoring how those parameters alter results based on their target organism, sequence depth, or mutation rate.
For example, using default gap opening and extension penalties in BLAST or Clustal Omega might be optimal for mammalian sequences but completely misleading when analyzing highly divergent viral genomes or ancient bacterial DNA strains. Similarly, when using tools like AlphaFold or PyMOL for structural proteomics, ignoring the micro-environmental variables or the resolution thresholds can render your structural predictions scientifically invalid. When academic workloads become overwhelming and you struggle to parameterise complex pipelines, it is often wise to enlist professional assistance; a reliable platform can help you do my assignment, ensuring that your scripts, parameter files, and analytical justifications are custom-tuned to your university’s exact marking rubrics.
Table 1: Strategic Breakdown of Student Failures vs. Computational Standards
| Project Domain | Common Student Mistake | Standard Professional Solution |
| Next-Gen Sequencing | Omitting adapter trimming and low-quality base filtering. | Execute Trimmomatic/Fastp workflows; enforce PHRED score thresholds > Q30. |
| Differential Expression | Using raw p-values without adjusting for multiple testing. | Apply Benjamini-Hochberg (FDR) adjustments within DESeq2 or EdgeR frameworks. |
| Phylogenetics | Selecting distance-based models (NJ) without bootstrapping. | Utilize Maximum Likelihood (IQ-TREE) with minimum 1,000 bootstrap replicates. |
| Structural Biology | Assuming predicted protein structures match active biological states. | Validate model via Ramachandran plots (PROCHECK) and molecular dynamics simulations. |
3. The Statistical Pitfall: Ignoring Multiple Testing Corrections and Sample Size
Bioinformatics is essentially large-scale data science applied to molecular biology. A typical transcriptomic study measures the expression levels of over 20,000 genes simultaneously across treatment and control groups. If a student applies a standard Student’s t-test with a conventional significance threshold of $\alpha = 0.05$ to each gene individually, they will encounter a massive statistical issue known as the False Discovery Rate (FDR).
At an alpha level of 0.05, purely by chance, 5% of 20,000 genes—meaning approximately 1,000 genes—will appear to be differentially expressed even if there is absolutely no biological difference between the samples. Students frequently build extensive biological interpretations around these false discoveries, detailing biochemical pathways that are nothing more than statistical artifacts. To achieve professional academic standards (EEAT), you must utilize rigorous multiple testing corrections, such as the Benjamini-Hochberg procedure or the Bonferroni correction, typically integrated within robust R packages like DESeq2 or edgeR.
4. Inadequate Code Documentation and Poor Reproducibility
A core pillar of scientific excellence in bioinformatics is reproducibility. If an assessor cannot take your repository, script, or workflow notebook and replicate your exact figures and output tables, your project fails fundamental scientific evaluation. Many biology-focused students treat coding as a temporary chore, writing monolithic scripts filled with hardcoded absolute file paths (e.g., C:/Users/Student/Desktop/Data/raw_files/) and completely un-annotated logic loops.
When this code is evaluated on an external machine or an institutional high-performance computing (HPC) cluster like Australia’s National Computational Infrastructure (NCI), the script crashes instantly due to missing path references, un-declared library dependencies, or version mismatches. To avoid losing substantial documentation marks, adhere to strict software development practices:
- Utilize relative file paths across all scripts.
- Document every function, parameter choice, and data step thoroughly.
- Maintain a clean configuration or environment file (e.g., a Conda environment.yml or Docker container definition) ensuring absolute transparency regarding package versions.
5. “The Biological Disconnect”: Interpreting Computational Outputs in Isolation
It is quite easy to get caught up in the pure data science aspects of bioinformatics—optimizing machine learning classification metrics, writing elegant bash scripts, or maximizing the speed of parallelized alignment pipelines. However, assessors frequently note that students run into trouble when they fail to link their data metrics back to actual biology. A machine learning model that achieves 98% accuracy in diagnosing a disease state based on a microarray dataset is functionally useless if it relies on features that are known extraction artifacts or unrelated housekeeping genes.
Bioinformatics is an interpretive science. Every clusters heatmap, PCA plot, or structural model must be contextualised within known metabolic path mechanisms, cellular localization dynamics, or evolutionary paradigms. If you observe an over-expression of a specific set of genes, map them to standard biological repositories like Gene Ontology (GO) or Kyoto Encyclopedia of Genes and Genomes (KEGG) to explain the biological mechanism driving the data pattern. Never let the computational tools speak without biological evaluation.
Frequently Asked Questions (FAQs)
Q1: Which programming language should I focus on for my bioinformatics project?
Python and R remain the foundational standards across Australian universities. Python is generally preferred for core structural modeling, sequence processing, pipeline integration, and machine learning workflows. R is highly specialized for complex statistical processing, differential expression analysis (DESeq2), and producing publication-grade data visualizations (ggplot2).
Q2: Why is the choice of reference genome so critical in alignment workflows?
Using an outdated or incorrect reference genome build (e.g., confusing hg19 with GRCh38 in human genetics) alters the chromosome coordinates of your aligned reads. This mismatch leads to incorrect structural variant assignments, invalidates downstream SNP annotations, and detaches your project from current genomic research databases.
Q3: How do I handle missing data in large biological datasets?
Missing data must be approached carefully. If the missing entries are minimal, you may utilize sophisticated imputation techniques like K-Nearest Neighbors (KNN) or Multivariate Imputation by Chained Equations (MICE). However, if a sample lacks comprehensive quality data across major loci, it should be entirely excluded during preprocessing to preserve the integrity of the analysis.
About the Author: Dr. Alistair Vance
Dr. Alistair Vance is a Senior Academic Consultant and Chief Bioinformatics Strategist at MyAssignmentHelp. He holds a PhD in Computational Genomics from the University of Queensland and has spent over nine years lecturing across major Australian biotechnology frameworks. Dr. Vance specializes in optimizing scalable NGS assembly pipelines and provides targeted instructional support to students conquering high-performance data analytics workflows.
Academic References & Data Sources
- Australian Bioinformatics and Computational Biology Society (ABACBS). (2025). Core Competencies and Curriculum Guidelines for Tertiary Bioinformatics Education in Australia. ABACBS Education Committee.
- Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., … & Mortazavi, A. (2016). A survey of best practices for RNA-seq data analysis. Genome Biology, 17(1), 13.
- National Computational Infrastructure (NCI) Australia. (2024). HPC Optimization for Genomic Analysis and High-Throughput Sequence Mapping pipelines. NCI Technical Reports.